Future Plans¶
Overview¶
VRS covers a fundamental subset of data types to represent variation, thus far predominantly related to the replacement of a subsequence in a reference sequence. Increasing its applicability will require supporting more complex types of variation, including:
genotypes
structural variation
mosaicism and chimerism
categorical variation
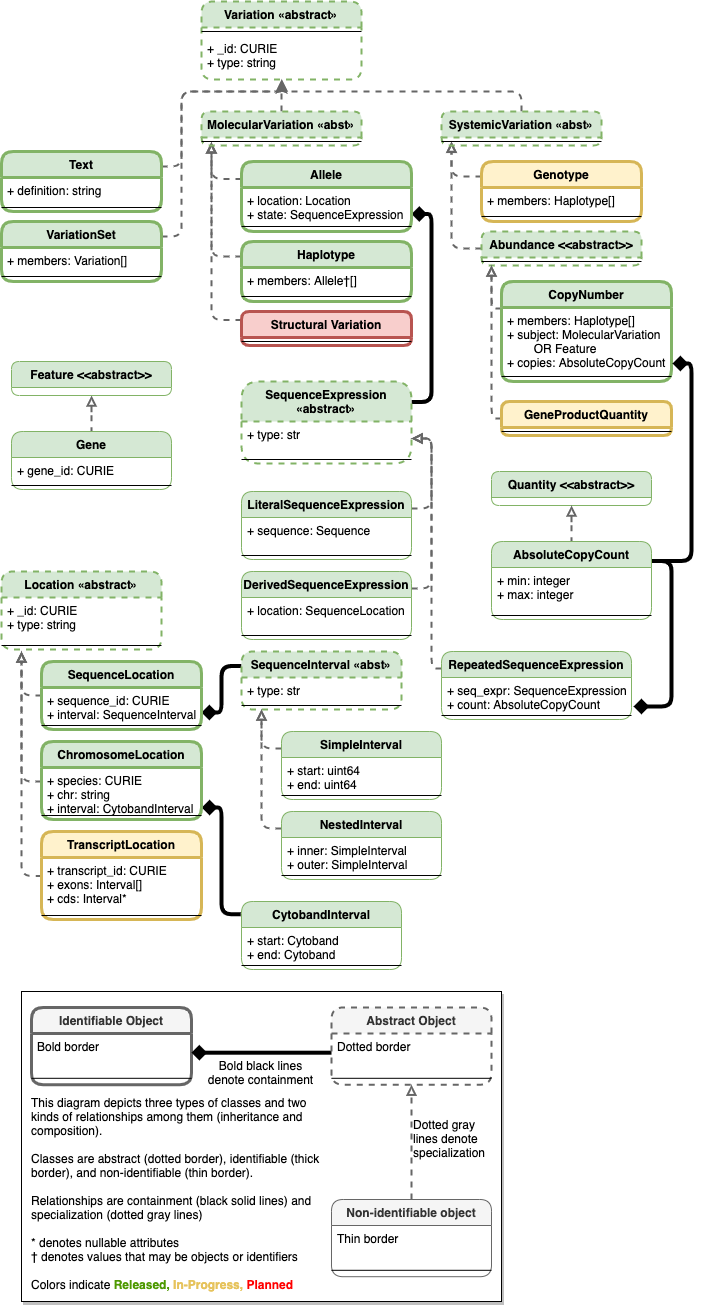
Planned Variation Representation Specfication Schema¶
See Current schema diagram for legend.
Existing classes are colored green. Components that are undergoing testing and evaluation and are candidates for the next release cycle are yellow. Components that are planned but still undergoing requirement gathering and initial development are colored red.
[source]
The following sections provide a preview of planned concepts under way to address a broader representation of variation.
Intervals and Locations¶
VRS uses Location subclasses to define where variation occurs. The schema is designed to be extensible to new kinds of Intervals and Locations in order to support, for example, fuzzy coordinates or feature-based locations.
ComplexInterval¶
Representation of complex coordinates based on relative locations or offsets from a known location. Examples include “left of” a given position and intronic positions measured from intron-exon junctions.
Computational definition
Under development.
Information model
Under development.
Variation Classes¶
Additional Variation concepts that are being planned for future consideration in the specification. See Variation for more information.
Structural Variation¶
Note
This concept is being refined. Please comment at https://github.com/ga4gh/vrs/issues/103
The aberrant joining of two segments of DNA that are not typically contiguous. In the context of joining two distinct coding sequences, translocations result in a gene fusion, which is also covered by this VRS definition.
Computational definition
A joining of two sequences is defined by two Location objects and an indication of the join “pattern” (advice needed on conventional terminology, if any).
Information model
Under consideration. See https://github.com/ga4gh/vrs/issues/28.
Examples
t(9;22)(q34;q11) in BCR-ABL
Categorical Variation¶
Some variations are defined by categorical concepts, rather than specific locations and states. These variations go by many terms, including categorical variants, bucket variants, container variants, or variant classes. These forms of variation are not described by any broadly-recognized variation format, but modeling them is a key requirement for the representation of aggregate variation descriptions as commonly found in biomedical literature. Our future work will focus on the formal specification for representing these variations with sets of rules, which we currently call Categorical Variation.