Future Plans¶
Overview¶
The VR-Spec covers a fundamental subset of data types to represent variation, thus far predominantly related to the replacement of a subsequence in a reference sequence. Increasing its applicability will require supporting more complex types of variation, including:
- alternative coordinate types such as nested ranges
- feature-based coordinates such as genes, cytogenetic bands, and exons
- haplotypes and genotypes
- copy number variation
- structural variation
- mosaicism and chimerism
- rule-based variation
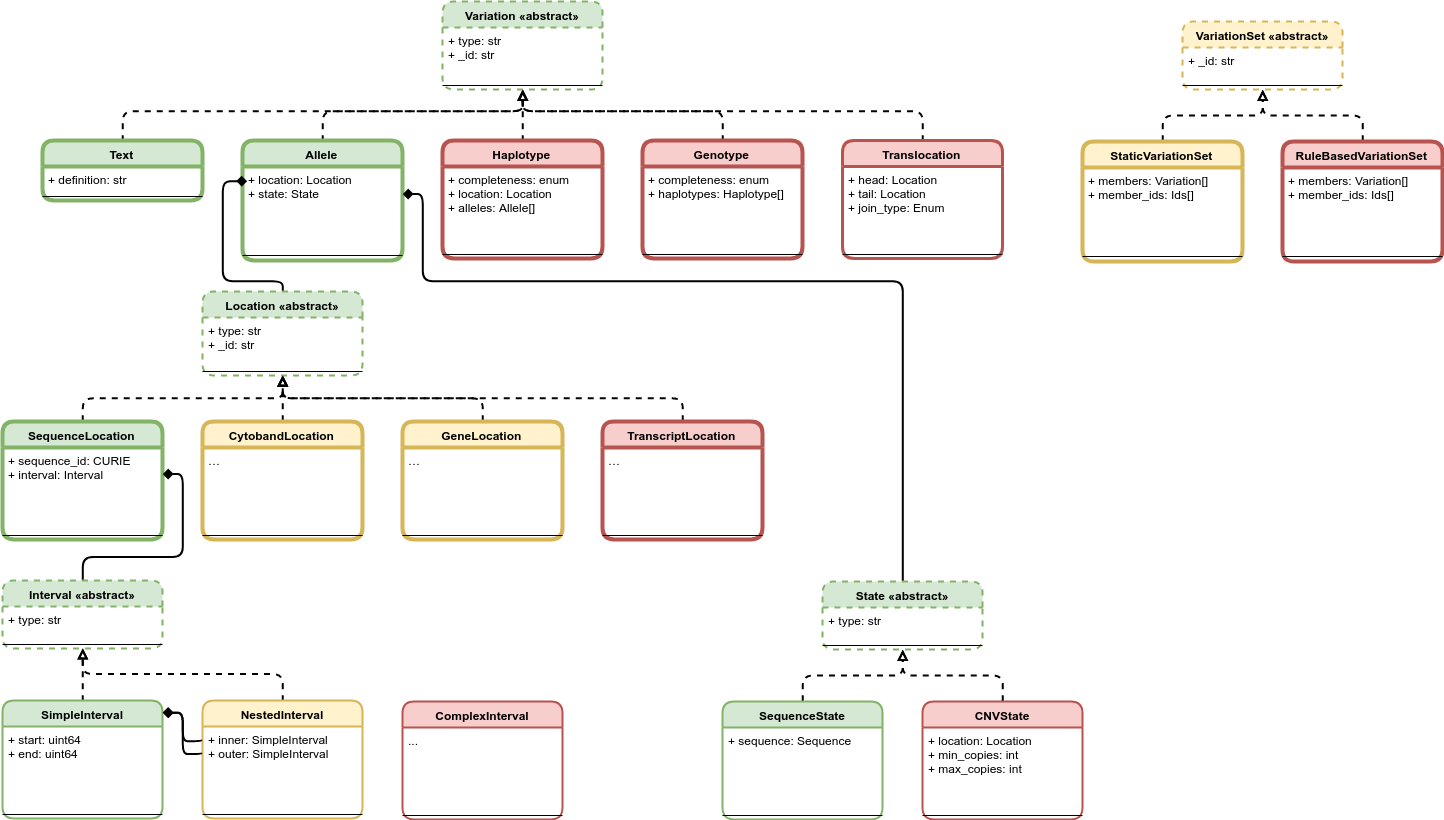
An illustration of planned components for the VR Schema. Version 1.0 components are colored green. Components that are undergoing testing and evaluation and are candidates for the next release cycle are colored yellow. Components that are planned but still undergoing requirement gathering and initial development are colored red. The VR Schema requires the use of multiple composite objects, which are grouped under four abstract classes: Variation, Location (Abstract Class), State (Abstract Class), and Interval (Abstract Class). These classes and their relationships to the representation of Variation are illustrated here. All classes have a string type. Dashed borders denote abstract classes. Abstract classes are not instantiated. Thin solid borders denote classes that may be instantiated but are not identifiable. Bold borders denote identifiable objects (i.e., may be serialized and identified by computed identifier). Solid arrow lines denoted inheritance. Subclasses inherit all attributes from their parent. Inherited attributes are not shown.
The following sections provide a preview of planned concepts under way to address a broader representation of variation.
Intervals and Locations¶
VR-Spec uses Interval (Abstract Class) and Location (Abstract Class) subclasses to define where variation occurs. The schema is designed to be extensible to new kinds of Intervals and Locations in order to support, for example, fuzzy coordinates or feature-based locations.
NestedInterval¶
Biological definition
None
Computational definition
An Interval (Abstract Class) comprised of an inner and outer SimpleInterval. The NestedInterval allows for the definition of “fuzzy” range endpoints by designating a potentially included region (the outer SimpleInterval) and required included region (the inner SimpleInterval).
Information model
| Field | Type | Limits | Description |
|---|---|---|---|
| type | string | 1..1 | Interval type; MUST be set to ‘NestedInterval’ |
| inner | SimpleInterval | 1..1 | known interval |
| outer | SimpleInterval | 1..1 | potential interval |
Implementation guidance
- Implementations MUST enforce values 0 ≤ outer.start ≤ inner.start ≤ inner.end ≤ outer.end. In the case of double-stranded DNA, this constraint holds even when a feature is on the complementary strand.
ComplexInterval¶
Biological definition
Representation of complex coordinates based on relative locations or offsets from a known location. Examples include “left of” a given position and intronic positions measured from intron-exon junctions.
Computational definition
Under development.
Information model
Under development.
CytobandLocation¶
Biological definition
Imprecise chromosomal locations based on chromosomal staining.
Computational definition
Cytogenetic bands are defined by a chromosome name, band, and sub-band. In VR-Spec, a cytogenetic location is an interval on a single chromsome with a start and end band and subband.
Information model
Under development.
GeneLocation¶
Biological definition
The symbolic location of a gene.
Computational definition
A gene location is made by reference to a gene identifier from NCBI, Ensembl, HGNC, or other public trusted authority.
Information model
| Field | Type | Limits | Description |
|---|---|---|---|
| _id | CURIE | 0..1 | Location Id; MUST be unique within document |
| type | string | 1..1 | Location type; MUST be set to ‘GeneLocation’ |
| gene_id | CURIE | 1..1 | CURIE-formatted gene identifier using NCBI numeric gene id. |
Notes
- gene_id MUST be specified as a CURIE, using a CURIE prefix of “NCBI” and CURIE reference with the numeric gene id. Other trusted authorities MAY be permitted in future releases.
Implementation guidance
- GeneLocations MAY be converted to SequenceLocation using external data. The source of such data and mechanism for implementation is not defined by this specification.
State Classes¶
Additional State (Abstract Class) concepts that are being planned for future consideration in the specification.
CNVState¶
Note
This concept is being refined. Please comment at https://github.com/ga4gh/vr-spec/issues/46.
Biological definition
Variations in the number of copies of a segment of DNA. Copy number variations cover copy losses or gains and at known or unknown locations (including tandem repeats). Variations MAY occur at precise SequenceLocations, within nested intervals, or at GeneLocations. There is no lower or upper bound on CNV sizes.
Computational definition
Under development.
Information model
| Field | Type | Limits | Description |
|---|---|---|---|
| type | string | 1..1 | State type; MUST be set to ‘CNVState’ |
| location | Location (Abstract Class) | 1..1 | the Location of the copy (‘null’ if unknown) |
| min_copies | int | 1..1 | The minimum number of copies |
| max_copies | int | 1..1 | The maximum number of copies |
Variation Classes¶
Additional Variation concepts that are being planned for future consideration in the specification. See Variation for more information.
Haplotypes¶
Biological definition
A specific combination of Alleles that occur together on single sequence in one or more individuals.
Computational definition
A specific combination of non-overlapping Alleles that co-occur on the same reference sequence.
Information model
| Field | Type | Limits | Description |
|---|---|---|---|
| _id | CURIE | 0..1 | Variation Id; MUST be unique within document |
| type | string | 1..1 | Variation type; MUST be set to ‘Haplotype’ |
| location | Location (Abstract Class) | 1..1 | Where Haplotype is located |
| completeness | enum | 1..1 | Declaration of completeness of the Haplotype definition. Values are:
|
| alleles | CURIE[] | 2..* | List of Alleles that comprise this Haplotype. |
Implementation guidance
- The Haplotype location (as specified by the location_id) MAY refer to a subsequence of the reference sequence, such as a subsequence of an entire chromosome.
- All Alleles in a Haplotype MUST be defined on the same reference sequence as specified by location_id.
- Alleles within a Haplotype MUST not intersect (“intersect” is defined in SimpleInterval).
- All Location Intervals are to be interpreted in the context of the underlying reference sequence, irrespective of insertions or deletions by other “upstream” Alleles within the Haplotype.
- When reporting an Haplotype, completeness MUST be set according to
these criteria:
- “COMPLETE” only if the entire reference sequence was assayed and all in-phase Alleles are reported in this Haplotype.
- “PARTIAL” only if the entire reference sequence was assayed, other in-phase Alleles exist, and are NOT reported in this Haplotype. This is an assertion of unreported variation.
- “UNKNOWN” otherwise. This value is the default and SHOULD be used if neither “COMPLETE” nor “PARTIAL” applies. These cases include, but are not limited to, assays that do not fully cover the reference sequence and an unwillingness by the reporter to declare the existence or absence of other in-phase Alleles.
- A Haplotype with an empty list of Alleles and completeness set to “COMPLETE” is an assertion of an unchanged reference sequence.
- When projecting a Haplotype from one sequence to a larger sequence, a “complete” Haplotype becomes an “unknown” Haplotype on the target sequence. Furthermore, this change is not reversible.
Notes
Alleles within a Haplotype are, by definition, “cis” or “in-phase”. (“In phase” and “cis” refer to features that exist on instances of covalently bonded sequences.)
Haplotypes are often given names, such as ApoE3 or A*33:01 for convenience.
- Examples: A*33:01:01 (IMGT/HLA)
When used to report Haplotypes, the completeness property enables data providers (e.g, diagnostic labs) to indicate that other Alleles exist, may exist, or do not exist. Data providers may not assay the full reference sequence or may withhold other in-phase Alleles in order to protect patient privacy.
When used to define Haplotypes, the completeness property enables implementations to permit (PARTIAL) or preclude (COMPLETE) the existence of other variation when matching a Haplotype to a set of observed Alleles.
Data consumers MAY wish to use the completeness property in order to provide accurate context for Allele interpretation or to select data used in association studies.
Sources
- ISOGG: Haplotype — A haplotype is a combination of alleles (DNA sequences) at different places ( loci) on the chromosome that are transmitted together. A haplotype MAY be one locus, several loci, or an entire chromosome depending on the number of recombination events that have occurred between a given set of loci.
- SO: haplotype (SO:0001024) — A haplotype is one of a set of coexisting sequence variants of a haplotype block.
- GENO: Haplotype (GENO:0000871) - A set of two or more sequence alterations on the same chromosomal strand that tend to be transmitted together.
Genotypes¶
Biological definition
The genetic state of an organism, whether complete (defined over the whole genome) or incomplete (defined over a subset of the genome).
Computational definition
A list of Haplotypes.
Information model
| Field | Type | Limits | Description |
|---|---|---|---|
| _id | CURIE | 0..1 | Variation Id; MUST be unique within document |
| type | string | 1..1 | Variation type; MUST be set to ‘Genotype’ |
| completeness | enum | 1..1 | Declaration of completeness of the Haplotype definition. Values are:
|
| haplotypes | CURIE[] | 2..* | List of Haplotypes; length MUST agree with ploidy of genomic region |
Implementation guidance
- Haplotypes in a Genotype MAY occur at different locations or on different reference sequences. For example, an individual may have haplotypes on two population-specific references.
- Haplotypes in a Genotype MAY contain differing numbers of Alleles or Alleles at different Locations.
Notes
- The term “genotype” has two, related definitions in common use. The narrower definition is a set of alleles observed at a single location and with a ploidy of two, such as a pair of single residue variants on an autosome. The broader, generalized definition is a set of alleles at multiple locations and/or with ploidy other than two.The VR-Spec Genotype entity is based on this broader definition.
- The term “diplotype” is often used to refer to two haplotypes. The VR-Spec Genotype entity subsumes the conventional definition of diplotype. Therefore, the VR-Spec model does not include an explicit entity for diplotypes. See this note for a discussion.
- The VR-SPec model makes no assumptions about ploidy of an organism or individual. The number of Haplotypes in a Genotype is the observed ploidy of the individual.
- In diploid organisms, there are typically two instances of each autosomal chromosome, and therefore two instances of sequence at a particular location. Thus, Genotypes will often list two Haplotypes. In the case of haploid chromosomes or haploinsufficiency, the Genotype consists of a single Haplotype.
- A consequence of the computational definition is that Haplotypes at overlapping or adjacent intervals MUST NOT be included in the same Genotype. However, two or more Alleles MAY always be rewritten as an equivalent Allele with a common sequence and interval context.
- The rationale for permitting Genotypes with Haplotypes defined on different reference sequences is to enable the accurate representation of segments of DNA with the most appropriate population-specific reference sequence.
Sources
SO: Genotype (SO:0001027) — A genotype is a variant genome, complete or incomplete.
Note
Genotypes represent Haplotypes with arbitrary ploidy The VR-Spec defines Haplotypes as a list of Alleles, and Genotypes as a list of Haplotypes. In essence, Haplotypes and Genotypes represent two distinct dimensions of containment: Haplotypes represent the “in phase” relationship of Alleles while Genotypes represents sets of Haplotypes of arbitrary ploidy.
There are two important consequences of these definitions: There is no single-location Genotype. Users of SNP data will be familiar with representations like rs7412 C/C, which indicates the diploid state at a position. In the VR-Spec, this is merely a special case of a Genotype with two Haplotypes, each of which is defined with only one Allele (the same Allele in this case). The VR-Spec does not define a diplotype type. A diplotype is a special case of a VR-Spec Genotype with exactly two Haplotypes. In practice, software data types that assume a ploidy of 2 make it very difficult to represent haploid states, copy number loss, and copy number gain, all of which occur when representing human data. In addition, assuming ploidy=2 makes software incompatible with organisms with other ploidy. The VR-Spec makes no assumptions about “normal” ploidy.
In other words, the VR-Spec does not represent single-position Genotypes or diplotypes because both concepts are subsumed by the Allele, Haplotype, and Genotypes entities.
Translocations¶
Note
This concept is being refined. Please comment at https://github.com/ga4gh/vr-spec/issues/103
Biological definition
The aberrant joining of two segments of DNA that are not typically contiguous. In the context of joining two distinct coding sequences, translocations result in a gene fusion, which is also covered by this VR-Spec definition.
Computational definition
A joining of two sequences is defined by two Location (Abstract Class) objects and an indication of the join “pattern” (advice needed on conventional terminology, if any).
Information model
Under consideration. See https://github.com/ga4gh/vr-spec/issues/28.
Examples
t(9;22)(q34;q11) in BCR-ABL
Rule-based Variation¶
Some variations are defined by categorical concepts, rather than specific locations and states. These variations go by many terms, including categorical variants, bucket variants, container variants, or variant classes. These forms of variation are not described by any broadly-recognized variation format, but modeling them is a key requirement for the representation of aggregate variation descriptions as commonly found in biomedical literature. Our future work will focus on the formal specification for representing these variations with sets of rules, which we currently call Rule-based Variation.
RuleLocation¶
RuleLocation is a subclass of Location (Abstract Class) intended to capture locations defined by rules instead of specific contiguous sequences. This includes locations defined by sequence characteristics, e.g. microsatellite regions.
RuleState¶
RuleState is a subclass of State (Abstract Class) intended to capture states defined by categorical rules instead of sequence states. This includes gain- / loss-of-function, oncogenic, and truncating variation.
Variation Sets¶
Note
The VR-Spec anticipates the need for sets of variation. Sets MAY be static (immutable) or dynamic (changeable), and might be defined manually, by an equivalence function, or by an expansion functions. Furthermore, equivalence and expansion functions might be user-defined. This concept is being refined. Please comment at https://github.com/ga4gh/vr-spec/issues/15